When a father and his two kids developed an array of peculiar symptoms, doctors, Brazilian researchers and the NIH partnered to find a diagnosis and path to treatment.
10:27 PM
Author |

Oxford, Michigan resident Stan Gibson inherited a rare disease from his father, who got it from his father.
They all had developed unusual symptoms throughout their lives, including childhood glaucoma, psoriasis-looking skin rashes, calcium build-up throughout the body and arthritis or spontaneous tendon rupture.
"My grandfather and some of my dad's siblings died young, and we didn't really know why," Stan recalled.
He was the only one of his three siblings to inherit the strange condition. "We didn't know too much, but my wife and I figured this condition must only be passed down to males."
He and his wife, Eryn, also thought that, based on the familial pattern of when the condition developed, their five sons were in the clear from having the disease since they didn't show any symptoms in early childhood.
"Everyone in Stan's family developed glaucoma between the ages of 3 and 5," Eryn said.
It wasn't until their then 3-year-old daughter, Rachel, started walking around holding her eyes and head, complaining of pain and light sensitivity, that alarm bells went off in the family and they rushed to find a doctor that might be able to give them more answers.
Recently moved to Michigan from California, the family didn't know where to go. But luckily, after many surgeries and no luck, the family found themselves at C.S. Mott Children's Hospital and the Kellogg Eye Center.
"It felt like being a Patient X"
All the surgeries had left Rachel's eyes severely damaged.
At one point, Rachel's iris, the colored ring around the pupil, had actually slipped out of the middle of her eye and went into her sclera, the white outer layer of the eye.
"She looked like she had a cat eye since the iris was such a strange shape," Eryn recalls.
But soon, the blue of Rachel's iris started coming up through the sclera and the panicked parents were advised to go to Kellogg Eye Center. There, Brenda Bohnsack, M.D., Ph.D. was able to reconstruct Rachel's eye and completely save her vision.
Rachel began seeing Bohnsack regularly. Because the family was impressed with the level of care Rachel was receiving, Stan also switched to Kellogg Eye Center and began care with glaucoma surgeon Sayoko Moroi, M.D., Ph.D., and cornea and refractive surgeon Shahzad Mian, M.D. He needed a repeat corneal transplant which was performed by Mian because of the high eye pressure that would cause his cornea to swell and his grafts to fail.
It wasn't until the Gibson family's then 13-year-old son Adam randomly proclaimed "I can't see anything" as they were returning home from a family outing that Bohnsack, Lev Prasov, M.D., Ph.D., an ophthalmic genetics specialist, and Julia Richards, Ph.D., a glaucoma geneticist at the Kellogg Eye Center, became more interested in the family's mysterious case of glaucoma and set out to try and learn more about this multi-system, genetic disease.
"It felt like being a Patient X," said Stan.
Not regular childhood glaucoma
"We thought Adam was in the clear since he was older than 6; we had stopped screening our children for the glaucoma," said Eryn. "But one day we got home and Adam stayed in the van. When we asked him what was wrong, he said he couldn't see anything. He went completely blind, just like that."
Like Podcasts? Add the Michigan Medicine News Break on iTunes, Google Podcast or anywhere you listen to podcasts.
Bohnsack, now a pediatric glaucoma surgeon and head of pediatric ophthalmology at Lurie Children's Hospital, got Adam in for an emergency surgery within hours. His eye pressure was over 50, according to the family. According to Prasov, a normal eye pressure is between 10-21 mm Hg. Thankfully, the emergency surgery saved most of Adam's vision, although he would live with a small amount of peripheral vision loss.
The strong family history and the peculiar symptoms led the Kellogg Eye Center glaucoma genetics team to dig deeper and look for a genetic cause that links the symptoms shared between Stan, Rachel and Adam. Ultimately, the answer came from genetic analysis through a collaboration between Prasov and the National Eye Institute of the National Institutes of Health (NIH), where he did his clinical fellowship.
Working with the NIH
The first step for the Kellogg Eye Center glaucoma team was meticulously interviewing family members and drawing a pedigree. The Gibson family even took a trip to Utah to get blood and saliva samples from family members and sent them to the researchers. They determined that this was an autosomal dominant condition, with a 50% risk of being passed down in the family.
In partnership with the NIH, Prasov and his team sequenced all of the protein coding DNA from affected and unaffected family members, a process called whole exome sequencing. After scouring through the data, they found a compelling misspelling in the DDX58 gene that had never been observed before. This gene encodes for an RNA binding protein involved in the immune system.
The team discovered Stan, Rachel and Adam all shared this mutant variant, and this was the eureka moment where all of the mysterious symptoms started to make sense.
"Rachel was resistant to light before we went to Kellogg Eye Center, but now there's just a delay in her adjustment from light to dark. For Adam, although he didn't tell us at first, he had headaches from a young age. Stan, besides the blindness, also has sensitive skin, unusual spots all over his body and tendons that tear easily," said Eryn.
Maybe one of the most peculiar symptoms of all, Rachel, Adam and Stan all had issues with their baby teeth because they weren't losing them naturally at the typical age children lose baby teeth.
Stan had to have his baby teeth taken out because his adult teeth were being impacted, and now at 11-years-old, Rachel is still losing baby teeth. Adam had issues with his teeth, but also his other bones, which is why he started seeing an orthopedic surgeon at 6-years-old.
If it wasn't for Kellogg Eye Center, both of my kids would be blind.Eryn Gibson
Eight years later Adam, now 21 and a gymnastics coach, would have high blood pressure and experience a carotid artery blowout. Thankfully, though, his neck muscles saved his life.
"The teeth abnormalities, as well as the carotid artery blowout, are likely due to the calcification in the body that patients with this illness can present," said Prasov. "With the genetic testing, we were able to conclude this disease was Singleton-Merten syndrome type II – an extremely rare, autosomal, dominant condition with only 3 families documented in literature."
With their collective expertise, the team was able to better characterize the family's symptoms: pediatric-onset glaucoma, spontaneous tendon rupture, arthritis, and a psoriasis-looking skin rash. Glaucoma and skin rash were the most prominent symptoms.
There weren't many cases in literature to compare the family's disease presentation to, so Prasov, whose research focuses on inherited eye diseases, worked with colleagues at the National Institutes of Health and U-M clinician-scientists Johann Gudjonsson, M.D., Ph.D., a dermatologist, and J. Michelle Kahlenberg, M.D., Ph.D., a rheumatologist, to more deeply phenotype the family.
"When looking for new genes, it can take a considerable amount of time. Once we could perform sequencing, we could move a lot faster to get a diagnosis. The Gibson family had an official diagnosis approximately a year after we performed the sequencing." said Prasov.
He adds that working in the field of genetics comes with certain challenges; the provider making the diagnosis wants to be absolutely certain that the genetic change is causing the disease, as it can have a big impact on clinical management.
More certainty came with the identification of another family across the globe in Belém, Brazil, that had a similar peculiar set of clinical findings and the same genetic variant in DDX58. By comparing notes among all of the identified families, Prasov and his team were able to give a definitive diagnosis.
But the final piece of this medical puzzle required additional work in the laboratory.
The study
According to Prasov, very few families with mutations in DDX58 have a multi-system disorder featuring calcification in the body, skin rashes and childhood glaucoma.
Published in the Journal of Medical Genetics, Prasov led an international team, including researchers and clinicians from U-M, the NIH, Brazil, and Harvard, to provide the first complete molecular and histological assessment of the eye and skin findings in Singleton-Merten syndrome type II. Together, they definitively showed that the family's mutation behaves similarly to other mutant proteins in how it sets off an inflammatory cascade in the body in absence of a definitive RNA trigger.
"Because of the communication between these doctors, more of this condition is now better understood," said Eryn. "Their curiosity led to answers, but also helped us feel like we were finally being heard and cared about. Finally, after generations of unanswered questions and enormous health challenges someone took this seriously and had access to the resources to find answers about this condition."
Since skin is the most available tissue for molecular analysis, Gudjonsson and his team were able to obtain samples from Stan and Adam to compare Stan's gene expression to his children. Somewhat surprisingly, they found that the inflammatory response was only active in affected skin, and not in neighboring skin or in the blood.
The study team also found that the family in Brazil had a more severe case of Singleton-Merten syndrome. By working with the Brazilian clinicians and researchers, Prasov was able to compare data and see if other genetic triggers or environmental influences affect the presentation of the disease.
"This is something I want to explore more," he said. "Rachel and Adam don't have many of the system effects of this disease, but they could develop them in the future. Understanding all the factors that may provoke more severe illness may help us be able to prevent those developments from occurring."
MORE FROM MICHIGAN: Sign up for our weekly newsletter
The study reinforces the idea that genetic triggers or environmental influences may affect the presentation of Singleton-Merten syndrome, as the research team found that some family members carrying the mutation only have the most common feature of childhood glaucoma, while others have more extensive disease features that affect the immune system.
"This means there could be other children in the world with this glaucoma that don't have the other features of the disease," said Prasov. "But they're at risk for developing those features later in life and suffer serious health consequences if providers don't know to look out for all these symptoms."
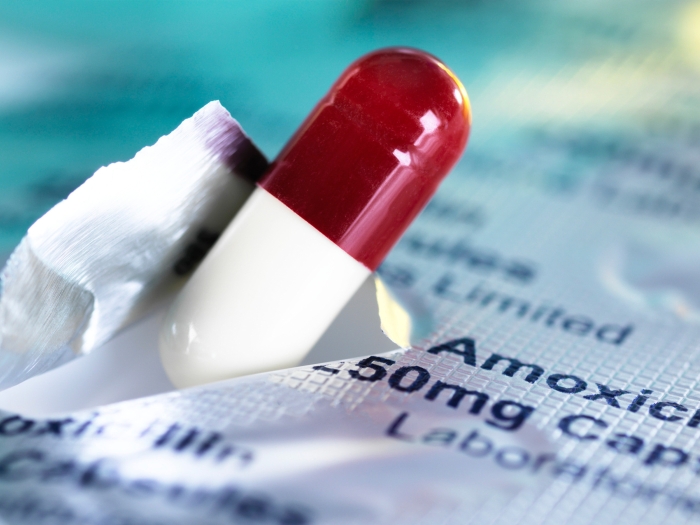
There are drugs like Rinvoq, a medication that Stan now takes, that can help calm the immune system and disrupt the downstream pathway associated with more serious disease. According to Stan, the medication has helped his skin discoloration resolve.
"I'm so grateful to know what this condition is. Knowing how to treat it would be a miracle, but you have to start somewhere," said Stan. "This research will benefit my own children but also their children when they start families of their own. They know what to look out for."
"This study highlights how a genetic diagnosis can have a big impact on clinical practice and patient outcomes," said Prasov. "Understanding this disease pathogenesis is also important in the field of glaucoma care and research." Prasov now leads a multidisciplinary clinic in which he partners with pediatric medical geneticists to provide ophthalmic genetic care for families like the Gibsons.
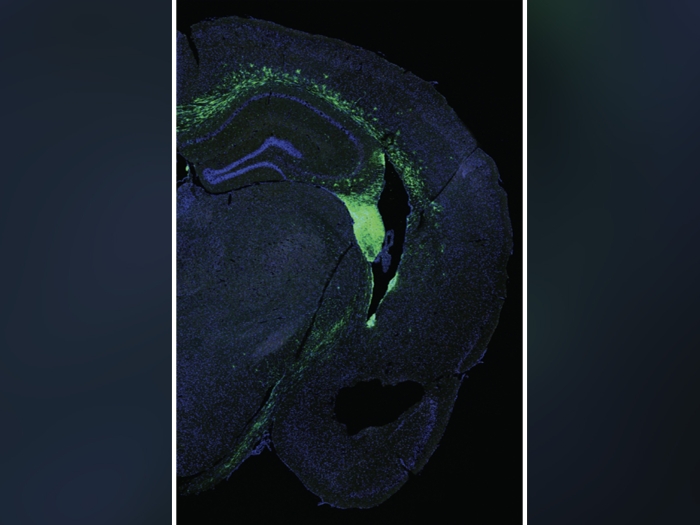
The next step for the U-M research team is to generate an animal model to better understand the disease and hopefully find a targeted drug treatment that may have fewer side effects. They also plan to further study the role of DDX58 in the eye, since the RNA binding protein can sense viral infections like Rubella and Zika: viruses known to provoke glaucoma. The hope is that they can find a way to block this receptor or the downstream signaling, in turn preventing the glaucoma from developing.
"Stan has a severe case of Singleton-Merten so my hope is that if researchers can crack his case, they can know how to treat anyone with the condition," said Eryn. "If it wasn't for Kellogg Eye Center, both of my kids would be blind. I'm just grateful beyond words."
This research was supported by the National Eye Institute (NEI K12 EY022299, NEI P30 EY07003, RBH, BG, and BPB), the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (R01-AR060802, P30-AR075043 and K01-AR072129), the National Institute of Allergy and Infectious Diseases (R01-AR069071), the A. Alfred Taubman Medical Research Institute, the Parfet Emerging Scholar Award and the Roche Postdoctoral Fellowship.
Paper cited: "DDX58(RIG-I)-related disease is associated with tissue-specific interferon pathway activation," Journal of Medical Genetics. DOI: 10.1136/jmedgenet-2020-10744

Explore a variety of healthcare news & stories by visiting the Health Lab home page for more articles.

Department of Communication at Michigan Medicine
Want top health & research news weekly? Sign up for Health Lab’s newsletters today!